Energy-Dispersive X-ray Spectrometry (EDS)
The methods described in this chapter are specific to the following signals:
EDSTEM
This chapter describes step-by-step the analysis of an EDS spectrum (SEM or TEM).
Note
See also the EDS tutorials.
Spectrum loading and parameters
The sample and data used in this section are described in [Burdet2013], and can be downloaded using:
>>> #Download the data (130MB)
>>> from urllib.request import urlretrieve, urlopen
>>> from zipfile import ZipFile
>>> files = urlretrieve("https://www.dropbox.com/s/s7cx92mfh2zvt3x/"
... "HyperSpy_demos_EDX_SEM_files.zip?raw=1",
... "./HyperSpy_demos_EDX_SEM_files.zip")
>>> with ZipFile("HyperSpy_demos_EDX_SEM_files.zip") as z:
>>> z.extractall()
Loading data
All data are loaded with the load() function, as described in
detail in Loading files. HyperSpy is able to import
different formats, among them “.msa” and “.rpl” (the raw format of Oxford
Instruments and Bruker).
Here are three examples of files exported by Oxford Instruments software (INCA). For a single spectrum:
>>> s = hs.load("Ni_superalloy_1pix.msa")
>>> s
<Signal1D, title: Signal1D, dimensions: (|1024)>
For a spectrum image (The .rpl file is recorded as an image in this example,
The method as_signal1D() set it back to a one
dimensional signal with the energy axis in first position):
>>> si = hs.load("Ni_superalloy_010.rpl").as_signal1D(0)
>>> si
<Signal1D, title: , dimensions: (256, 224|1024)>
Finally, for a stack of spectrum images, using “*” as a wildcard character:
>>> si4D = hs.load("Ni_superalloy_0*.rpl", stack=True)
>>> si4D = si4D.as_signal1D(0)
>>> si4D
<Signal1D, title:, dimensions: (256, 224, 2|1024)>
Microscope and detector parameters
First, the signal type (“EDS_TEM” or “EDS_SEM”) needs to be set with the
set_signal_type() method. By assigning the
class of the object, specific EDS methods are made available.
>>> s = hs.load("Ni_superalloy_1pix.msa")
>>> s.set_signal_type("EDS_SEM")
>>> s
<EDSSEMSpectrum, title: Signal1D, dimensions: (|1024)>
You can also specify the signal type as an argument of
the load() function:
>>> s = hs.load("Ni_superalloy_1pix.msa", signal_type="EDS_SEM")
>>> s
<EDSSEMSpectrum, title: Signal1D, dimensions: (|1024)>
HyperSpy will automatically load any existing microscope parameters from the
file, and store them in the metadata
attribute (see Metadata structure). These parameters can be displayed
as follows:
>>> s = hs.load("Ni_superalloy_1pix.msa", signal_type="EDS_SEM")
>>> s.metadata.Acquisition_instrument.SEM
├── Detector
│ └── EDS
│ ├── azimuth_angle = 63.0
│ ├── elevation_angle = 35.0
│ ├── energy_resolution_MnKa = 130.0
│ ├── live_time = 0.006855
│ └── real_time = 0.0
├── beam_current = 0.0
├── beam_energy = 15.0
└── tilt_stage = 38.0
You can also set these parameters directly:
>>> s = hs.load("Ni_superalloy_1pix.msa", signal_type="EDS_SEM")
>>> s.metadata.Acquisition_instrument.SEM.beam_energy = 30
or by using the
set_microscope_parameters() method:
>>> s = hs.load("Ni_superalloy_1pix.msa", signal_type="EDS_SEM")
>>> s.set_microscope_parameters(beam_energy = 30)
or through the GUI:
>>> s = hs.load("Ni_superalloy_1pix.msa", signal_type="EDS_SEM")
>>> s.set_microscope_parameters()
EDS microscope parameters preferences window
Any microscope and detector parameters that are not found in the imported file
will be set by default. These default values can be changed in the
Preferences class (see preferences).
>>> hs.preferences.EDS.eds_detector_elevation = 37
or through the GUI:
>>> hs.preferences.gui()
EDS preferences window
Energy axis
The size, scale and units of the energy axis are automatically imported from
the imported file, where they exist. These properties can also be set
or adjusted manually with the AxesManager
(see Axis properties for more info):
>>> si = hs.load("Ni_superalloy_010.rpl",
... signal_type="EDS_TEM").as_signal1D(0)
>>> si.axes_manager[-1].name = 'E'
>>> si.axes_manager['E'].units = 'keV'
>>> si.axes_manager['E'].scale = 0.01
>>> si.axes_manager['E'].offset = -0.1
or through the GUI:
>>> si.axes_manager.gui()
Axis properties window
Copying spectrum calibration
All of the above parameters can be copied from one spectrum to another
with the get_calibration_from()
method.
>>> # s1pixel contains all the parameters
>>> s1pixel = hs.load("Ni_superalloy_1pix.msa", signal_type="EDS_TEM")
>>>
>>> # si contains no parameters
>>> si = hs.load("Ni_superalloy_010.rpl",
... signal_type="EDS_TEM").as_signal1D(0)
>>>
>>> # Copy all the properties of s1pixel to si
>>> si.get_calibration_from(s1pixel)
Describing the sample
The description of the sample is also stored in the
metadata attribute. It can be displayed using:
>>> s = hs.datasets.example_signals.EDS_TEM_Spectrum()
>>> s.add_lines()
>>> s.metadata.Sample.thickness = 100
>>> s.metadata.Sample
├── description = FePt bimetallic nanoparticles
├── elements = ['Fe', 'Pt']
├── thickness = 100
└── xray_lines = ['Fe_Ka', 'Pt_La']
The following methods are either called “set” or “add”.
“set” methods overwrite previously defined values
“add” methods add to the previously defined values
Elements
The elements present in the sample can be defined using the
set_elements() and
add_elements() methods. Only element
abbreviations are accepted:
>>> s = hs.datasets.example_signals.EDS_TEM_Spectrum()
>>> s.set_elements(['Fe', 'Pt'])
>>> s.add_elements(['Cu'])
>>> s.metadata.Sample
└── elements = ['Cu', 'Fe', 'Pt']
X-ray lines
Similarly, the X-ray lines can be defined using the
set_lines() and
add_lines() methods. The corresponding
elements will be added automatically.
Several lines per element can be defined at once.
>>> s = hs.datasets.example_signals.EDS_TEM_Spectrum()
>>> s.set_elements(['Fe', 'Pt'])
>>> s.set_lines(['Fe_Ka', 'Pt_La'])
>>> s.add_lines(['Fe_La'])
>>> s.metadata.Sample
├── elements = ['Fe', 'Pt']
└── xray_lines = ['Fe_Ka', 'Fe_La', 'Pt_La']
The X-ray lines can also be defined automatically, if the beam energy is set. The most excited X-ray line is selected per element (highest energy above an overvoltage of 2 (< beam energy / 2)):
>>> s = hs.datasets.example_signals.EDS_SEM_Spectrum()
>>> s.set_elements(['Al', 'Cu', 'Mn'])
>>> s.set_microscope_parameters(beam_energy=30)
>>> s.add_lines()
>>> s.metadata.Sample
├── elements = ['Al', 'Cu', 'Mn']
└── xray_lines = ['Al_Ka', 'Cu_Ka', 'Mn_Ka']
>>> s.set_microscope_parameters(beam_energy=10)
>>> s.set_lines([])
>>> s.metadata.Sample
├── elements = ['Al', 'Cu', 'Mn']
└── xray_lines = ['Al_Ka', 'Cu_La', 'Mn_La']
A warning is raised if you try to set an X-ray line higher than the beam energy:
>>> s = hs.datasets.example_signals.EDS_SEM_Spectrum()
>>> s.set_elements(['Mn'])
>>> s.set_microscope_parameters(beam_energy=5)
>>> s.add_lines(['Mn_Ka'])
Warning: Mn Ka is above the data energy range.
Elemental database
HyperSpy includes an elemental database, which contains the energy of the X-ray lines.
>>> hs.material.elements.Fe.General_properties
├── Z = 26
├── atomic_weight = 55.845
└── name = iron
>>> hs.material.elements.Fe.Physical_properties
└── density (g/cm^3) = 7.874
>>> hs.material.elements.Fe.Atomic_properties.Xray_lines
├── Ka
│ ├── energy (keV) = 6.404
│ └── weight = 1.0
├── Kb
│ ├── energy (keV) = 7.0568
│ └── weight = 0.1272
├── La
│ ├── energy (keV) = 0.705
│ └── weight = 1.0
├── Lb3
│ ├── energy (keV) = 0.792
│ └── weight = 0.02448
├── Ll
│ ├── energy (keV) = 0.615
│ └── weight = 0.3086
└── Ln
├── energy (keV) = 0.62799
└── weight = 0.12525
Finding elements from energy
To find the nearest X-ray line for a given energy, use the utility function
get_xray_lines_near_energy() to search the elemental
database:
>>> s = hs.datasets.example_signals.EDS_SEM_Spectrum()
>>> P = s.find_peaks1D_ohaver(maxpeakn=1)[0]
>>> hs.eds.get_xray_lines_near_energy(P['position'], only_lines=['a', 'b'])
['C_Ka', 'Ca_La', 'B_Ka']
The lines are returned in order of distance from the specified energy, and can be limited by additional, optional arguments.
Plotting
You can visualize an EDS spectrum using the
plot() method (see
visualisation). For example:
>>> s = hs.datasets.example_signals.EDS_SEM_Spectrum()
>>> s.plot()
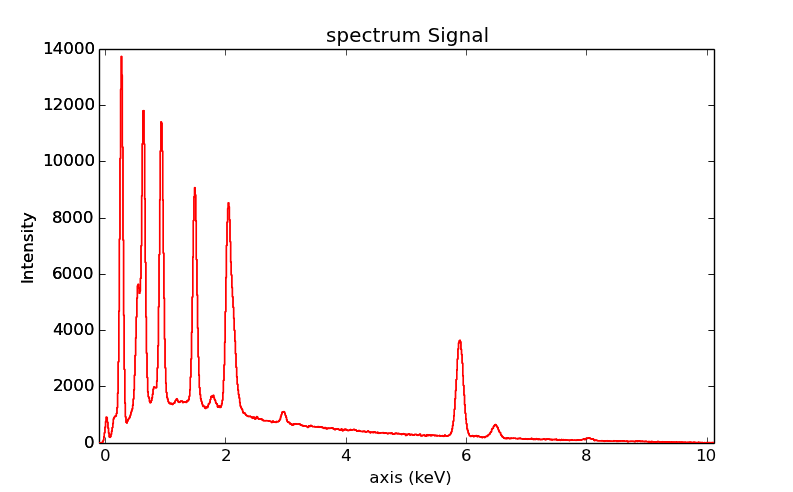
EDS spectrum
An example of multi-dimensional EDS data (e.g. 3D SEM-EDS) is given in visualisation multi-dimension.
Plotting X-ray lines
X-ray lines can be added as plot labels with
plot(). The lines are either retrieved
from metadata.Sample.Xray_lines, or selected with the same method as
add_lines() using the elements in
metadata.Sample.elements.
>>> s = hs.datasets.example_signals.EDS_SEM_Spectrum()
>>> s.add_elements(['C','Mn','Cu','Al','Zr'])
>>> s.plot(True)
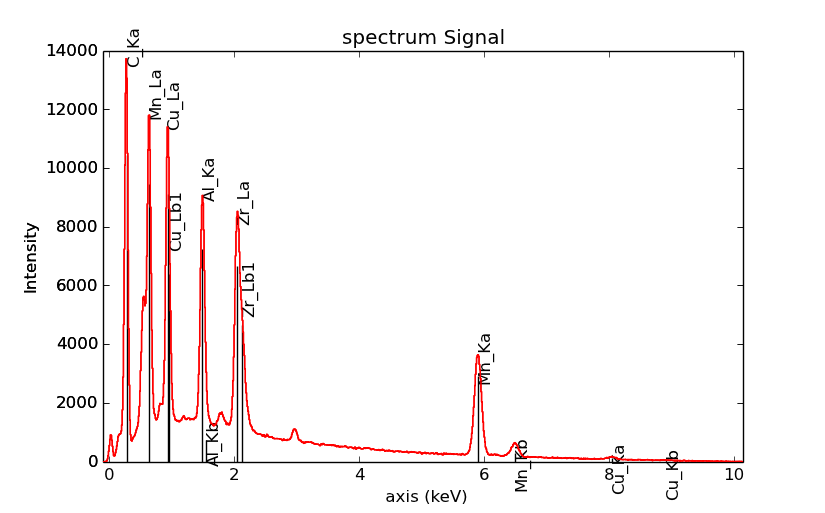
EDS spectrum plot with line markers
You can also select a subset of lines to label:
>>> s = hs.datasets.example_signals.EDS_SEM_Spectrum()
>>> s.add_elements(['C','Mn','Cu','Al','Zr'])
>>> s.plot(True, only_lines=['Ka','b'])
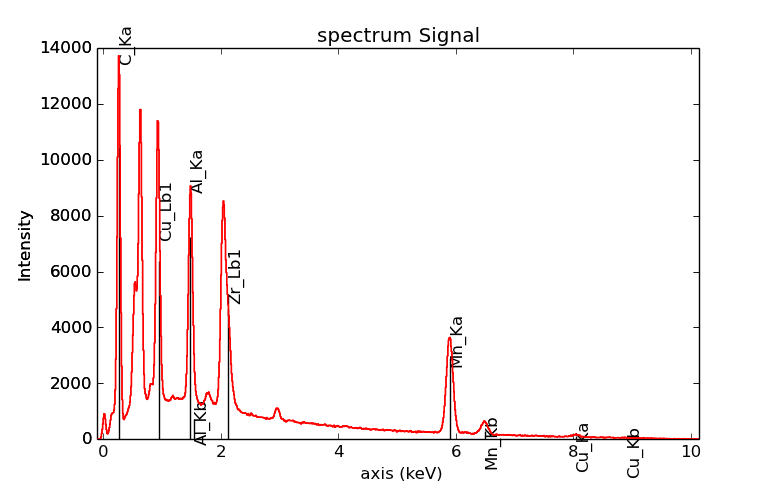
EDS spectrum plot with a selection of line markers
Getting the intensity of an X-ray line
The sample and data used in this section are described in [Rossouw2015], and can be downloaded using:
>>> #Download the data (1MB)
>>> from urllib.request import urlretrieve, urlopen
>>> from zipfile import ZipFile
>>> files = urlretrieve("https://www.dropbox.com/s/ecdlgwxjq04m5mx/"
... "HyperSpy_demos_EDS_TEM_files.zip?raw=1",
... "./HyperSpy_demos_EDX_TEM_files.zip")
>>> with ZipFile("HyperSpy_demos_EDX_TEM_files.zip") as z:
>>> z.extractall()
The width of integration is defined by extending the energy resolution of Mn Ka to the peak energy (energy_resolution_MnKa in the metadata):
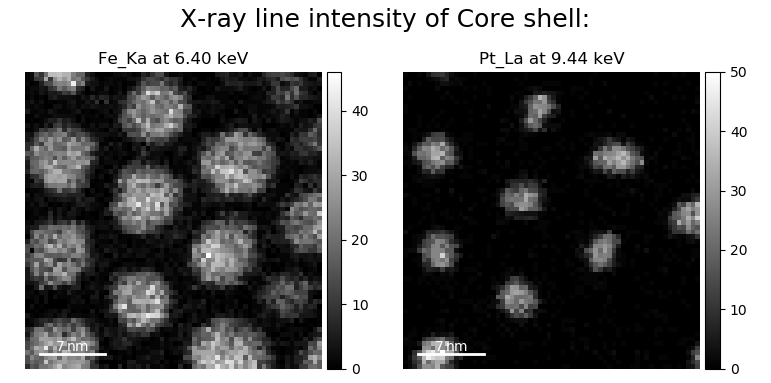
>>> s = hs.load('core_shell.hdf5')
>>> s.get_lines_intensity(['Fe_Ka'], plot_result=True)

Iron map as computed and displayed by get_lines_intensity
The X-ray lines defined in metadata.Sample.Xray_lines are used by default.
The EDS maps can be plotted using plot_images(), see plotting several images
for more information in setting plotting parameters.
>>> s = hs.load('core_shell.hdf5')
>>> s.metadata.Sample
├── elements = ['Fe', 'Pt']
└── xray_lines =['Fe_Ka', 'Pt_La']
>>> eds_maps = s.get_lines_intensity()
>>> hs.plot.plot_images(eds_maps, axes_decor='off', scalebar='all')
Finally, the windows of integration can be visualised using
plot() method:
>>> s = hs.datasets.example_signals.EDS_TEM_Spectrum().isig[5.:13.]
>>> s.add_lines()
>>> s.plot(integration_windows='auto')
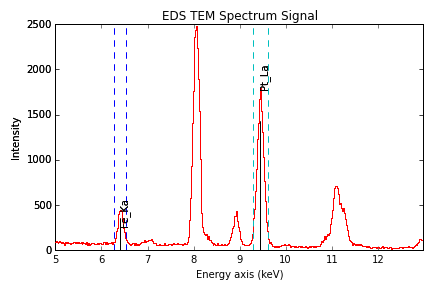
EDS spectrum with integration windows markers
Background subtraction
The background can be subtracted from the X-ray intensities with
get_lines_intensity().
The background value is obtained by averaging the intensity in two
windows on each side of the X-ray line.
The position of the windows can be estimated using
estimate_background_windows(), and
can be plotted using plot():
>>> s = hs.datasets.example_signals.EDS_TEM_Spectrum().isig[5.:13.]
>>> s.add_lines()
>>> bw = s.estimate_background_windows(line_width=[5.0, 2.0])
>>> s.plot(background_windows=bw)
>>> s.get_lines_intensity(background_windows=bw, plot_result=True)
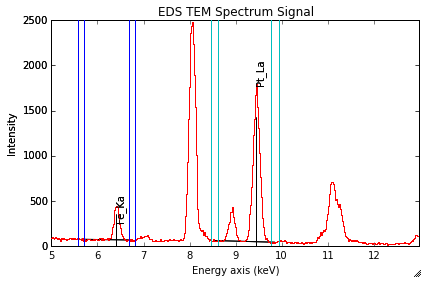
EDS spectrum with background subtraction markers.
EDS curve fitting
The intensity of X-ray lines can be extracted using curve-fitting in HyperSpy. This example uses an EDS-SEM spectrum of a test material (EDS-TM001) provided by BAM.
First, we load the spectrum, define the chemical composition of the sample and set the beam energy:
>>> s = hs.load('bam.msa')
>>> s.add_elements(['Al', 'Ar', 'C', 'Cu', 'Mn', 'Zr'])
>>> s.set_microscope_parameters(beam_energy=10)
Next, the model is created with
create_model(). One Gaussian is
automatically created per X-ray line, along with a polynomial for the
background.
>>> m = s.create_model()
>>> m.print_current_values()
Components Parameter Value
Al_Ka
A 65241.4
Al_Kb
Ar_Ka
A 3136.88
Ar_Kb
C_Ka
A 79258.9
Cu_Ka
A 1640.8
Cu_Kb
Cu_La
A 74032.6
Cu_Lb1
Cu_Ln
Cu_Ll
Cu_Lb3
Mn_Ka
A 47796.6
Mn_Kb
Mn_La
A 73665.7
Mn_Ln
Mn_Ll
Mn_Lb3
Zr_La
A 68703.8
Zr_Lb1
Zr_Lb2
Zr_Ln
Zr_Lg3
Zr_Ll
Zr_Lg1
Zr_Lb3
background_order_6
The width and the energies are fixed, while the heights of the sub-X-ray lines are linked to the main X-ray lines (alpha lines). The model can now be fitted:
>>> m.fit()
The background fitting can be improved with
fit_background() by enabling only energy
ranges containing no X-ray lines:
>>> m.fit_background()
The width of the X-ray lines is defined from the energy resolution (FWHM at
Mn Ka) provided by energy_resolution_MnKa in metadata. This parameter
can be calibrated by fitting with
calibrate_energy_axis():
>>> m.calibrate_energy_axis(calibrate='resolution')
Energy resolution (FWHM at Mn Ka) changed from 130.000000 to 131.927922 eV
Fine-tuning of specific X-ray lines can be achieved using
calibrate_xray_lines():
>>> m.calibrate_xray_lines('energy', ['Ar_Ka'], bound=10)
>>> m.calibrate_xray_lines('width', ['Ar_Ka'], bound=10)
>>> m.calibrate_xray_lines('sub_weight', ['Mn_La'], bound=10)
The result of the fit is obtained with the
get_lines_intensity() method.
>>> result = m.get_lines_intensity(plot_result=True)
Al_Ka at 1.4865 keV : Intensity = 65241.42
Ar_Ka at 2.9577 keV : Intensity = 3136.88
C_Ka at 0.2774 keV : Intensity = 79258.95
Cu_Ka at 8.0478 keV : Intensity = 1640.80
Cu_La at 0.9295 keV : Intensity = 74032.56
Mn_Ka at 5.8987 keV : Intensity = 47796.57
Mn_La at 0.63316 keV : Intensity = 73665.70
Zr_La at 2.0423 keV : Intensity = 68703.75
Finally, we visualize the result:
>>> m.plot()
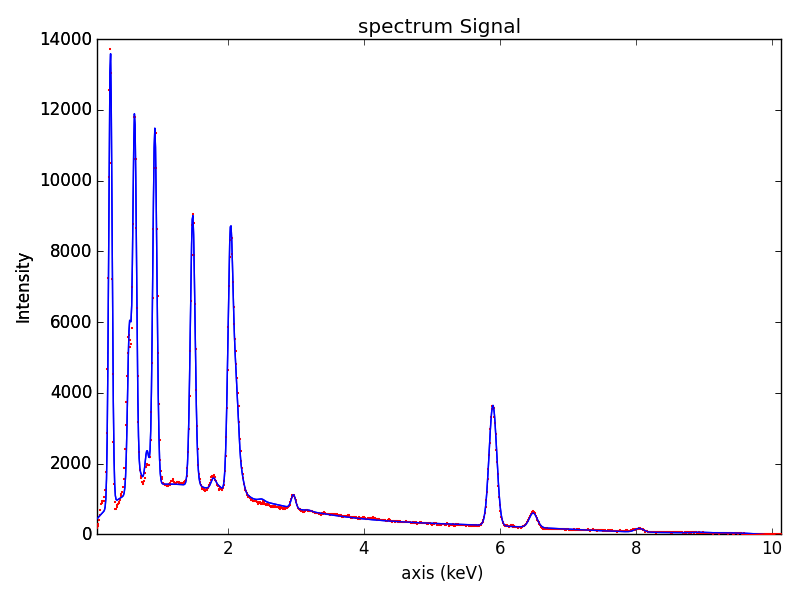
The following methods can be used to enable/disable different functionalities of X-ray lines when fitting:
EDS Quantification
HyperSpy includes three methods for EDS quantification with or without absorption correction:
Cliff-Lorimer
Zeta-factors
Ionization cross sections
Quantification must be applied to the background-subtracted intensities, which
can be found using get_lines_intensity().
The quantification of these intensities can then be calculated using
quantification().
The quantification method needs be specified as either ‘CL’, ‘zeta’, or ‘cross_section’. If no method is specified, the function will raise an exception.
A list of factors or cross sections should be supplied in the same order as
the listed intensities (please note that HyperSpy intensities in
get_lines_intensity() are in alphabetical
order).
A set of k-factors can be usually found in the EDS manufacturer software although determination from standard samples for the particular instrument used is usually preferable. In the case of zeta-factors and cross sections, these must be determined experimentally using standards.
Zeta-factors should be provided in units of kg/m^2. The method is described
further in [Watanabe1996]
and [Watanabe2006]. Cross sections should be
provided in units of barns (b). Further details on the cross section method can
be found in [MacArthur2016]. Conversion between
zeta-factors and cross sections is possible using
edx_cross_section_to_zeta() or
zeta_to_edx_cross_section().
Using the Cliff-Lorimer method as an example, quantification can be carried out as follows:
>>> s = hs.datasets.example_signals.EDS_TEM_Spectrum()
>>> s.add_lines()
>>> kfactors = [1.450226, 5.075602] #For Fe Ka and Pt La
>>> bw = s.estimate_background_windows(line_width=[5.0, 2.0])
>>> intensities = s.get_lines_intensity(background_windows=bw)
>>> atomic_percent = s.quantification(intensities, method='CL',
... factors=kfactors)
Fe (Fe_Ka): Composition = 15.41 atomic percent
Pt (Pt_La): Composition = 84.59 atomic percent
The obtained composition is in atomic percent, by default. However, it can be
transformed into weight percent either with the option
quantification():
>>> # With s, intensities and kfactors from before
>>> s.quantification(intensities, method='CL', factors=kfactors,
... composition_units='weight')
Fe (Fe_Ka): Composition = 4.96 weight percent
Pt (Pt_La): Composition = 95.04 weight percent
or using atomic_to_weight():
>>> # With atomic_percent from before
>>> weight_percent = hs.material.atomic_to_weight(atomic_percent)
The reverse method is weight_to_atomic().
The zeta-factor method needs both the ‘beam_current’ (in nA) and the acquisition or dwell time (referred to as ‘real_time’ in seconds) in order to obtain an accurate quantification. Both of the these parameters can be assigned to the metadata using:
>>> s.set_microscope_parameters(beam_current=0.5)
>>> s.set_microscope_parameters(real_time=1.5)
If these parameters are not set, the code will produce an error. The zeta-factor method will produce two sets of results. Index [0] contains the composition maps for each element in atomic percent, and index [1] contains the mass-thickness map.
The cross section method needs the ‘beam_current’, dwell time (‘real_time’) and probe area in order to obtain an accurate quantification. The ‘beam_current’ and ‘real_time’ can be set as shown above. The ‘probe_area’ (in nm^2) can be defined in two different ways.
If the probe diameter is narrower than the pixel width, then the probe is being under-sampled and an estimation of the probe area needs to be used. This can be added to the metadata with:
>>> s.set_microscope_parameters(probe_area=0.00125)
Alternatively, if sub-pixel scanning is used (or the spectrum map was recorded at a high spatial sampling and subsequently binned into much larger pixels) then the illumination area becomes the pixel area of the spectrum image. This is a much more accurate approach for quantitative EDS and should be used where possible. The pixel width could either be added to the metadata by putting the pixel area in as the ‘probe_area’ (above) or by calibrating the spectrum image (see Setting axis properties).
Either approach will provide an illumination area for the cross_section quantification. If the pixel width is not set, the code will still run with the default value of 1 nm with a warning message to remind the user that this is the case.
The cross section method will produce two sets of results. Index [0] contains the composition maps for each element in atomic percent and index [1] is the number of atoms per pixel for each element.
Note
Please note that the function does not assume square pixels, so both the x and y pixel dimensions must be set. For quantification of line scans, rather than spectrum images, the pixel area should be added to the metadata as above.
Absorption Correction
Absorption correction can be included into any of the three quantification
methods by adding the parameter absorption_correction=True to the function.
By default, the function iterates the quantification function until a
tolerance value of 0.5% up to a maximum number of iterations is reached. The
maximum number of iterations is set to 30 by default, but can be increased by
specifying max_iterations in the function call. However, typically for TEM
experiments convergence is achieved after less then 5 iterations.
For example:
>>> s.quantification(intensities, method='cross_section',
... factors=factors, absorption_correction=True)
However for the kfactor method the user must additionally provide a sample thickness (in nm) either as a single float value or as a numpy array with the same dimensions as the navigation axes. If this is done the calculated mass_thickness is additionally outputted from the function as well as the composition maps for each element.
>>> s.quantification(intensities, method='CL',
... factors=factors, absorption_correction=True
... thickness=100.)
At this stage absorption correction is only applicable for parallel-sided, thin-film samples. Absorption correction is calculated on a pixel by pixel basis after having determined a sample mass-thickness map. It therefore may be a source of error in particularly inhomogeneous specimens.
Absorption correction can also only be applied to spectra from a single EDS detector. For systems that consist of multiple detectors, such as the Thermo Fisher Super-X, it is therefore necessary to load the spectra from each detector separately.
Utils
Mass absorption coefficient database
A mass absorption coefficient database [Chantler2005] is available:
>>> hs.material.mass_absorption_coefficient(
... element='Al', energies=['C_Ka','Al_Ka'])
array([ 26330.38933818, 372.02616732])
>>> hs.material.mass_absorption_mixture(
... elements=['Al','Zn'], weight_percent=[50,50], energies='Al_Ka')
2587.4161643905127
Electron and X-ray range
The electron and X-ray range in a bulk material can be estimated with
hs.eds.electron_range() and hs.eds.xray_range()
To calculate the X-ray range of Cu Ka in pure Copper at 30 kV in micron:
>>> hs.eds.xray_range('Cu_Ka', 30.)
1.9361716759499248
To calculate the X-ray range of Cu Ka in pure Carbon at 30kV in micron:
>>> hs.eds.xray_range('Cu_Ka', 30., hs.material.elements.C.
... Physical_properties.density_gcm3)
7.6418811280855454
To calculate the electron range in pure Copper at 30 kV in micron
>>> hs.eds.electron_range('Cu', 30.)
2.8766744984001607